

Idiopathic Pulmonary Fibrosis (IPF): Existing Challenges and New Frontiers
Executive Summary
- IPF is a chronic lung disease characterized by irreversible scarring and lung fibrosis, leading to decreased lung function over time.
- Prompt diagnosis is crucial for early intervention, but challenges in recognizing IPF often delay diagnosis until irreversible lung damage occurs.
- Only nintedanib and pirfenidone are approved by the Food and Drug Administration (FDA) for IPF treatment but they do not improve the quality of life or the survival of patients with IPF.
- Ongoing research focuses on novel therapies, most notably phosphodiesterase (PDE) inhibitors, offering new hope for individuals affected by this devastating, incurable disease.
What Is IPF?
IPF is an incurable, chronic, progressive lung disease characterized by scarring (fibrosis) in the lungs.1,2 Although the causes of IPF remain unclear, there is a consensus that lung epithelial injury initiates a process wherein fibroblasts are converted into myofibroblasts, and extracellular matrix components, such as collagen, accumulate.3 As IPF progresses, fibrotic tissue formation makes the lungs stiff and decreases their ability to expand and contract.1,2 This loss of elasticity over time causes a progressive decline in lung function.1,2
Primary symptoms of IPF include shortness of breath (dyspnea), a wet or dry cough, and eventually respiratory failure.1,2 Other symptoms may occur as lung function declines, including fatigue, weakness, weight loss, chest discomfort, tightness, and declining exercise tolerance.1,2 IPF carries with it a risk of complications and a high mortality rate, with the median survival time after diagnosis ranging from 3 to 5 years.1 For comparison, the median survival time of patients with chronic obstructive pulmonary disease (COPD) ranges from approximately 7 to 17 years, depending on disease severity.4
Therefore, improving the care of patients with IPF requires advances in diagnosis, management, and treatment.
Challenges With Diagnosis
Diagnosis Is Often Delayed
Typically, IPF is diagnosed in people aged 50 years and older.5 However, many patients report having had respiratory symptoms for up to 5 years before diagnosis; therefore, younger patients are likely underrepresented in prevalence estimates.6 Given that prompt intervention offers patients with IPF the best chance to minimize or slow irreparable lung damage, a delay in diagnosis can negatively affect prognosis.7-9 Early diagnosis and intervention will not cure IPF but may significantly preserve patient quality of life and extend life expectancy.7-9
In addition, IPF may not even be suspected until significant damage has occurred. Symptoms of IPF, such as cough and shortness of breath, are not specific to IPF, and this overlap in presentation with more common respiratory conditions (eg, asthma, COPD) complicates the differential diagnosis.7-9 Given the gradual onset of IPF, patients may not present to a physician for evaluation because they attribute mild respiratory symptoms and discomfort to aging or other causes.7-9 As a result, medical attention is often delayed until symptoms worsen and irreversible lung damage becomes apparent; thus, opportunities to slow IPF progression have been lost.6
Diagnosis May Require Invasive Testing
According to joint guidelines from the American Thoracic Society, European Respiratory Society, Japanese Respiratory Society, and Latin American Thoracic Association (ATS/ERS/JRS/ALAT), a diagnosis of IPF can be established with high-resolution computed tomography (HRCT) alone in patients presenting with subpleural/basal-predominant usual interstitial pneumonia and honeycombing with or without traction bronchiectasis/bronchiolectasis.7-9 However, patients who do not exhibit these definitive radiologic features may require an invasive surgical lung biopsy.7-9
The diagnostic value of a lung biopsy must be balanced against any associated risks, along with patient preferences and potential reluctance to undergo an invasive procedure.7-9 However, lung biopsy remains necessary if honeycombing is absent, even if all other pertinent features are present, or if radiologic features inconsistent with IPF are detected.7-9
Challenges With Management
Limited Treatment Options Are Available
Treatment options remain limited, although numerous therapies have been evaluated in prospective trials, including (1) a three-drug combination of N-acetylcysteine, a glucocorticoid, and azathioprine, (2) the tyrosine kinase inhibitor, imatinib, and (3) the nonselective endothelin receptor antagonist, bosentan.1,2,7 Based on trial results, guidelines for the management of IPF generally recommend against each of these options because they are ineffective.7 In fact, the three-drug combination is not only considered ineffective but also potentially harmful for patients due to long-term glucocorticoid exposure.1,2,7
Currently, the US FDA has approved only two agents for treating IPF: the tyrosine kinase inhibitor, nintedanib, and pirfenidone, an antifibrotic drug with pleiotropic effects.1,2,9 Nintedanib inhibits multiple tyrosine kinases, resulting in the inhibition of downstream signaling pathways implicated in fibroblast proliferation and the formation of scar tissue.1,2 Pirfenidone, on the other hand, has both anti-inflammatory and antifibrotic activity, but its exact mechanism is unknown.1,2 Neither nintedanib nor pirfenidone cures nor reverses the fibrotic processes in IPF.3
Based on this, the ATS/ERS/JRS/ALAT algorithm for the management of IPF includes only nintedanib and pirfenidone as recommended pharmacologic therapies (Figure 1).9
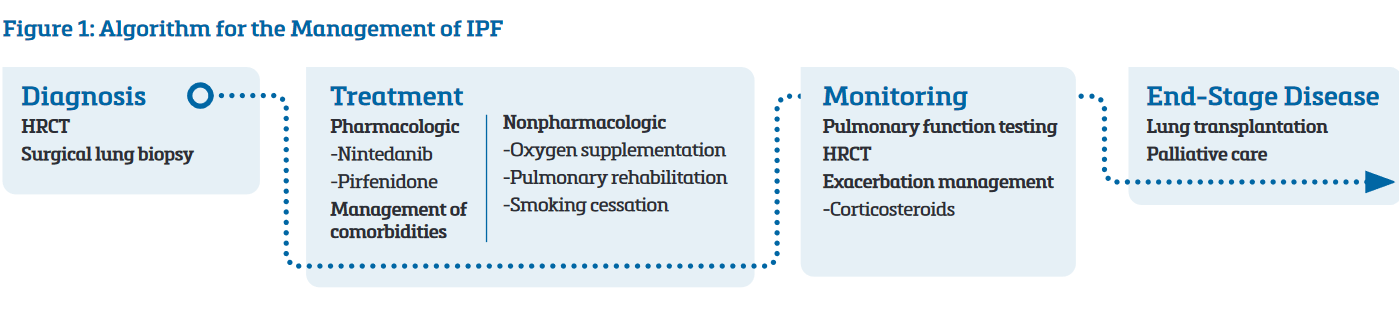
Treatment Relies Heavily on Nonpharmacologic Interventions
With limited pharmacologic options for IPF, treatment relies heavily on nonpharmacologic interventions such as pulmonary rehabilitation, supplemental oxygen, and smoking cessation (Figure 1).7,9 Lung transplantation may be an option in end-stage IPF, potentially improving symptoms, extending survival, and, in certain patients, slowing or halting disease progression (Figure 1).10 However, the criteria for selecting transplant-eligible patients are complex, and the procedure may not be suitable for many patients, such as older individuals who make up a substantial proportion of patients with end-stage disease.10 Even transplant-eligible patients derive only modest benefits from transplantation, as only two-thirds of transplant recipients live 3 years, and just over half live 5 years.2
Challenges With Drug Development
Few Drug Candidates Are in Late-Stage Development
Among investigational agents, only two drug classes have reached late-phase clinical development: pamrevlumab, a monoclonal antibody targeting connective tissue growth factor (CTGF), and BI 1015550, a PDE4B inhibitor (Figure 2).3,11
The anti-CTGF monoclonal antibody showed efficacy in a 2014 phase 3 trial.3,11 However, positive results were not replicated in two subsequent phase 3 trials, and development for this indication was terminated because the drug failed to meet its primary endpoint.12
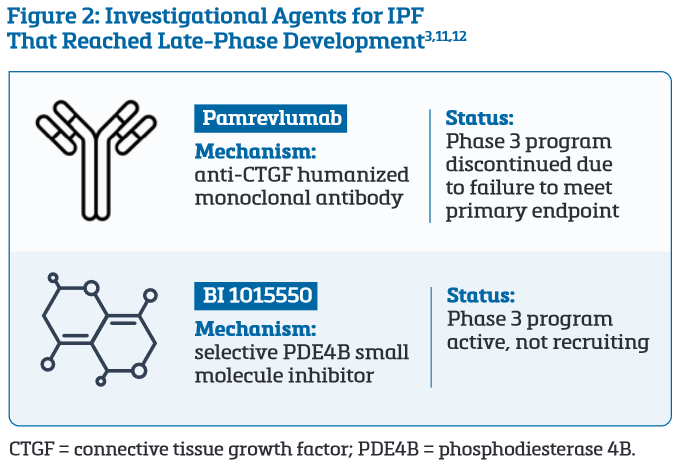
PDE Inhibitors Show Promise in the Treatment of IPF
Rationale For the Use of PDE Inhibitors
The cyclic nucleotides cAMP (cyclic adenosine 3’,5’-monophosphate) and cGMP (cyclic guanosine 3’,5’-monophosphate) play an important role in intracellular communication, and dysregulated cAMP and cGMP signaling is hypothesized to contribute to the fibrotic changes seen in IPF.13 PDE inhibitors prevent PDE from breaking down the phosphodiester bonds within cAMP and cGMP, which modifies the downstream signaling of these messenger molecules and subsequently reduces fibrosis.13
The anti-inflammatory effects of PDE inhibitors are well known, and potential antifibrotic effects are being studied.13 In preclinical studies, PDE inhibitors have been shown to prevent and reverse fibrosis, reduce collagen synthesis, and decrease the expression of several proinflammatory molecules in lung tissue.13 For example, roflumilast, a nonspecific PDE inhibitor, reduced levels of tumor necrosis factor-α (TNF-α), interleukin-13, transforming growth factor-β, and mucin 5ac in bronchoalveolar lavage fluid while also preventing the infiltration of neutrophils and macrophages.13
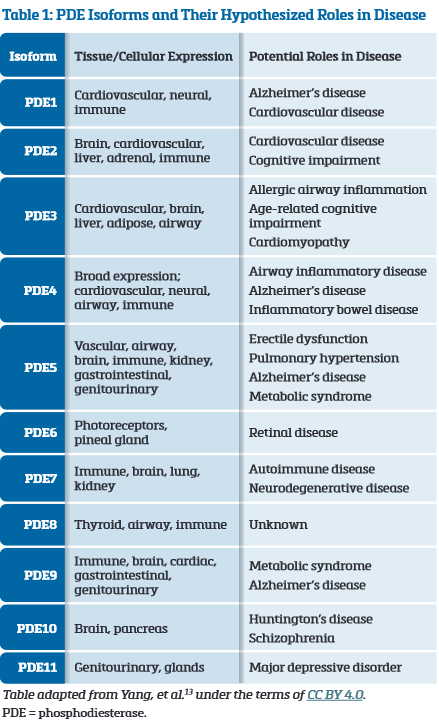
Selecting the Optimal PDE Isoform to Inhibit
PDE includes 11 isoforms with distinct structures, distributions, and functions (Table 1).13 PDE4, PDE7, and PDE8 specifically degrade cAMP, while PDE5, PDE6, and PDE9 degrade cGMP.13 Most cells contain multiple PDE isoforms in varying amounts.13 PDE4 is widely expressed in human lung tissue, and accordingly, PDE4 inhibitors are being studied as potential treatments for IPF as well as other airway inflammatory diseases (eg, asthma, COPD).13
PDE4 can be further divided into four subtypes: PDE4A, PDE4B, PDE4C, and PDE4D.13 In human primary lung fibroblasts, PDE4A, 4B, and 4D are abundantly expressed, whereas PDE4C is minimally expressed or not at all present.13 Of the PDE4 subtypes, gene silencing of PDE4B resulted in the greatest reduction in overall PDE4 enzyme activity.13 Consequently, PDE4B inhibitors appear to offer the greatest utility for preventing cAMP degradation in lung tissue.13
Moreover, tolerability issues are expected to limit the utility of nonspecific oral PDE4 inhibitors, with side effects that include gastrointestinal (GI) symptoms, headache, weight loss, and psychiatric symptoms.14 It has been proposed that adverse events, particularly GI events, may be associated with inhibition of the PDE4D subtype.13
Considering both potency and potential toxicity, PDE4B inhibition has been chosen as the most appropriate method of PDE4 inhibition in patients with IPF.13
Evidence to Support PDE4B Inhibition for the Treatment of IPF
Evidence From Preclinical Studies
In preclinical research, PDE4B inhibition preferentially inhibited the breakdown of cAMP by PDE4B, with downstream reduction of inflammation and fibrosis.15 PDE4B inhibition suppressed the release of proinflammatory molecules from purified human peripheral blood mononuclear cells.15 Additionally, ex vivo experiments in mice demonstrated the efficacy of PDE4B inhibition in reducing the release of the profibrotic molecule TNF-α in response to inflammatory stimuli.15
In animal models, PDE4B inhibition reduced neutrophil infiltration into bronchoalveolar lavage fluid, further supporting its potential anti-inflammatory effects in vivo.15 Moreover, PDE4B inhibition demonstrated therapeutic efficacy in murine models of lung fibrosis.15 Notably, PDE4B inhibition did not differ from controls in the production of nausea, and it produced substantially less nausea than the nonspecific PDE4 inhibitor roflumilast.15
The potential synergistic effect of combining a PDE4B inhibitor with an existing IPF therapy has also been explored.15 In several preclinical assays, nintedanib did not perform well when used alone; however, the combination of a PDE4B inhibitor with nintedanib exhibited synergistic improvements.15 Combining a PDE4B inhibitor with pirfenidone did not yield any additional effects beyond PDE4B inhibition alone.15 Collectively, these results highlight the potential of PDE4B inhibition in patients with IPF, both as monotherapy and as an adjunct to existing therapies.
Evidence From Phase 1 Clinical Studies
The pharmacokinetics, safety, and tolerability of an experimental PDE4B inhibitor were investigated in two separate phase 1 studies, one in healthy subjects and one in patients with IPF.16 In healthy volunteers, plasma drug concentrations increased rapidly, reaching peak concentrations of 1.25 to 1.52 hours after single and multiple oral doses.16 Drug concentrations then declined, with a terminal half-life of 16 to 27 hours.16 No significant differences in pharmacokinetic parameters were observed between healthy subjects and patients with IPF.16
PDE4B inhibition exhibited acceptable safety and tolerability in both healthy subjects and patients with IPF.16 GI upset and headache, common adverse events with nonselective PDE4 inhibitors, were also the most common adverse events with a PDE4B-selective agent.16
Evidence From Phase 2 Clinical Studies
In a double-blind, placebo-controlled phase 2 trial, the effectiveness and safety of an experimental PDE4B inhibitor were examined in 147 patients with IPF.17 Patients were randomly assigned to receive a PDE4B inhibitor (18 mg twice daily) or twice-daily placebo.17 The primary efficacy endpoint was a change in forced vital capacity (FVC) at 12 weeks, analyzed separately for patients with and without background antifibrotic treatment.17 Because this was an exploratory trial, inference testing was not performed to determine statistical significance.17
Patients without background antifibrotic treatment showed a median change in FVC of +5.7 mL with a PDE4B inhibitor compared to -81.7 mL with placebo.17 For patients on background antifibrotic therapy, the median changes were +2.7 mL with a PDE4B inhibitor and -59.2 mL with placebo.17 A Bayesian analysis demonstrated the superiority of active treatment over placebo, with a probability exceeding 0.98.17 Similar results were obtained using repeated measures analysis.17
The incidence of adverse events was higher in the PDE4B inhibitor group compared to placebo, regardless of background antifibrotic use.17 GI adverse events were the most common, including abdominal distension/pain, constipation, diarrhea, and nausea.17 Among patients without background antifibrotic use, 27% in the PDE4B inhibitor group and 16% in the placebo group reported GI adverse events.17 Among patients on background antifibrotic therapy, 37% in the PDE4B inhibitor group and 32% in the placebo group reported GI events.17 Diarrhea was the primary reason for PDE4B inhibitor treatment discontinuation (n=3) and was more prevalent in the PDE4B inhibitor group.17 Severe adverse events occurred in 4% of patients without background antifibrotic use in both treatment groups, in 6% of patients in the PDE4B inhibitor group with antifibrotic use, and in 20% of patients in the placebo group with antifibrotic use.17 Two fatal events occurred in the PDE4B inhibitor group: one due to COVID-19 pneumonia and one due to suspected vasculitis and IPF exacerbation.17 No adverse events related to depression or suicidal behavior were reported, but one patient in the PDE4B inhibitor group experienced suicidal ideation.17
Phase 3 Clinical Studies Are Ongoing
Based on the promising results from early-phase clinical development, a PDE4B inhibitor has advanced to phase-3 development.18 In a placebo-controlled, double-blind trial, patients with IPF are randomized in a 1:1:1 ratio to receive one of two doses of a PDE4B inhibitor or placebo twice daily for 52 weeks.18 Patients will be stratified by use of background antifibrotic treatment (nintedanib or pirfenidone).18 The primary endpoint is the absolute change in FVC at week 52.18 A key secondary endpoint is a composite of time to first acute IPF exacerbation, hospitalization due to respiratory cause, or death throughout the trial.18
Conclusion
IPF continues to present significant challenges with diagnosis and management due to its insidious onset, heterogeneous presentation, and absence of disease-modifying treatment options. Nonetheless, recent advances in drug development—particularly the continued development of PDE4B inhibitors—may address underlying profibrotic and inflammatory mechanisms in IPF to improve the outlook for this patient population.
Preclinical studies demonstrated the potential of PDE4B inhibition to mitigate inflammation and fibrosis, while early-phase clinical trials showed encouraging results in preserving lung function in patients with IPF. Phase 3 trial results that more fully elucidate the safety and efficacy profile of PDE4B inhibition are eagerly anticipated.
References:
- Barratt SL, Creamer A, Hayton C, Chaudhuri N. Idiopathic pulmonary fibrosis (IPF): an overview. J Clin Med. 2018;7(8):201. doi:10.3390/jcm7080201
- Lederer DJ, Martinez FJ. Idiopathic pulmonary fibrosis. N Engl J Med. 2018;3781811-1823. doi:10.1056/NEJMra1705751
- Guo H, Sun J, Zhang S, Nie Y, Zhou S, Zeng Y. Progress in understanding and treating idiopathic pulmonary fibrosis: recent insights and emerging therapies. Front Pharmacol. 2023;14:1205948. doi:10.3389/fphar.2023.1205948
- Leivseth L, Brumpton BM, Nilsen TI, Mai XM, Johnsen R, Langhammer A. GOLD classifications and mortality in chronic obstructive pulmonary disease: the HUNT Study, Norway. Thorax. 2013;68(10):914-921. doi:10.1136/thoraxjnl-2013-203270
- Esposito DB, Lanes S, Donneyong M, et al. Idiopathic pulmonary fibrosis in United States automated claims incidence, prevalence, and algorithm validation. Am Respir Crit Care Med. 2015;192(10):1200-7. doi:10.1164/rccm.201504-0818OC
- Hewson T, McKeever TM, Gibson JE, Navaratnam V, Hubbard RB, Hutchinson JP. Timing of onset of symptoms in people with idiopathic pulmonary fibrosis. Thorax. 2017:thoraxjnl-2017-210177. doi:10.1136/thoraxjnl-2017-210177
- Raghu G, Collard HR, Egan JJ, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183(6):788-824. doi:10.1164/rccm.2009-040GL
- Raghu G, Remy-Jardin M, Myers JL, et al. Diagnosis of idiopathic pulmonary fibrosis an official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. 2018;198(5):e44-e68. doi:10.1164/rccm.201807-1255ST
- Raghu G, Remy-Jardin M, Richeldi L, et al. Idiopathic pulmonary fibrosis (an update) and progressive pulmonary fibrosis in adults: an official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. 2022;205(9):e18-e47. doi:10.1164/rccm.202202-0399ST
- Leard LE, Holm AM, Valapour M, et al. Consensus document for the selection of lung transplant candidates: An update from the International Society for Heart and Lung Transplantation. J Heart Lung Transplant. 2021;40(11):1349-1379. doi:10.1016/j.healun.2021.07.005
- Sgalla G, Flore M, Siciliano M, Richeldi. Antibody-based therapies for idiopathic pulmonary fibrosis. Expert Opin Biol Ther. 2020;20(7):779-786. doi:10.1080/14712598.2020.1735346
- FibroGen announces topline results from phase 3 ZEPHYRUS-1 study of pamrevlumab for the treatment of idiopathic pulmonary fibrosis. Press release. FibroGen, Inc. June 26, 2023. Accessed April 22, 2024. https://investor.fibrogen.com/news-releases/news-release-details/fibrogen-announces-topline-results-phase-3-zephyrus-1-study
- Yang X, Xu Z, Hu S, Shen J. Perspectives of PDE inhibitor on treating idiopathic pulmonary fibrosis. Front Pharmacol. 2023;14:1111393. doi:10.3389/fphar.2023.1111393
- Phillips JE. Inhaled Phosphodiesterase 4 (PDE4) inhibitors for inflammatory respiratory diseases. Front Pharmacol. 2020;11:259. doi:10.3389/fphar.2020.00259
- Herrmann FE, Hesslinger C, Wollin L, et al. BI 1015550 is a PDE4B inhibitor and a clinical drug candidate for the oral treatment of idiopathic pulmonary fibrosis. Front Pharmacol. 2022;13:838449. doi:10.3389/fphar.2022.838449
- Maher TM, Schlecker C, Luedtke D, Bossert S, Zoz DF, Schultz TM. Phase I studies of BI 1015550, a preferential phosphodiesterase 4B inhibitor, in healthy males and patients with idiopathic pulmonary fibrosis. ERJ Open Res. 2022;8(4):00240-2022. doi:10.1183/23120541.00240-2022
- Richeldi L, Azuma A, Cottin V, et al. Trial of a preferential phosphodiesterase 4B inhibitor for idiopathic pulmonary fibrosis. N Engl J Med. 2022;386(23):2178-2187. doi:10.1056/NEJMoa2201737
- Richeldi L, Azuma A, Cottin V, et al. Design of a phase III, double-blind, randomised, placebo-controlled trial of BI 1015550 in patients with idiopathic pulmonary fibrosis (FIBRONEER-IPF). BMJ Open Respir Res. 2023;10(1):e001563. doi:10.1136/bmjresp-2022-001563



